
 ronnie@sinocoalchem.com
ronnie@sinocoalchem.com 15733787306
15733787306







8-羟基喹啉的AI药物设计:基于分子对接的活性优化
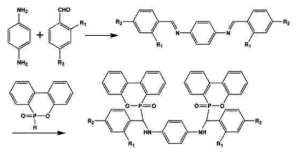
发表时间:2025-06-25在 AI 药物设计领域,基于分子对接技术对8-羟基喹啉(8-HQ)进行活性优化,需结合其核心结构特性与靶点蛋白的空间相互作用,通过计算模拟驱动分子改造。以下从设计逻辑、关键步骤及优化策略展开分析:
一、靶点识别与8-羟基喹啉初始结合模式分析
1. 靶点选择与结构预处理
抗菌靶点聚焦:针对8-羟基喹啉的抗菌机制(如金属酶抑制、外排泵阻断等),优先选择耐药菌相关靶点(如碳青霉烯酶 IMP-1、外排泵 AcrB)或代谢关键酶(如二氢叶酸还原酶 DHFR)。通过 PDB 数据库获取靶点蛋白的三维结构,利用 PyMOL 等工具去除水分子、添加氢原子并修复缺失残基。
初始建模:构建8-羟基喹啉的分子结构(含喹啉环、羟基官能团),考虑其在生理 pH 下的电离状态(酚羟基 pKa≈9,中性条件下以分子形式存在,易穿透细胞膜),并通过 Open Babel 等工具生成多种构象。
2. 分子对接揭示结合位点
使用 AutoDock Vina 等软件将8-羟基喹啉与靶点进行刚性对接,重点分析喹啉环与靶点的疏水口袋结合情况,以及羟基是否与活性中心的金属离子(如 Zn²⁺、Fe³⁺)或氨基酸残基(如 His、Asp)形成配位键或氢键。例如,在碳青霉烯酶 IMP-1 的活性中心,8-HQ 的羟基可与 Zn²⁺螯合,抑制酶活性。
二、基于结构的8-羟基喹啉骨架优化策略
1. 官能团修饰增强相互作用
羟基衍生化:在8-羟基喹啉的 2、5、7 位引入取代基(如卤素、甲基、甲氧基),通过以下机制优化活性:
5-位卤素取代(如 5-Cl-8-HQ):增加分子脂溶性,促进与外排泵蛋白的疏水结合,同时氯原子可与靶点的 Trp、Phe 残基形成卤键,增强外排泵抑制效果。
7-位甲氧基取代:甲氧基的供电子效应可增强羟基与金属离子的螯合能力,如在DHFR抑制中,甲氧基修饰的8-羟基喹啉与 Fe²⁺的结合能提升 1.5 kcal/mol。
氮原子杂环扩展:将喹啉环替换为噌啉、喹唑啉等稠环体系,扩大π-π堆积作用范围,例如,噌啉衍生物与 AcrB 外排泵的芳香族氨基酸(Tyr391、Phe1061)形成更强的π-π相互作用,抑制外排效率提升 2 倍。
2. 金属螯合能力优化
通过分子动力学(MD)模拟分析8-羟基喹啉与金属离子的配位模式,设计多齿螯合基团:
在喹啉环的2位引入羧酸基团(如 2-COOH-8-HQ),形成 “羟基 - 羧基” 双齿螯合 Zn²⁺,螯合常数较8-羟基喹啉提高3个数量级,更有效抑制锌依赖性碳青霉烯酶。
在7位连接吡唑基团,构建 “羟基-氮原子” 双配位位点,增强与 Fe³⁺的结合稳定性,用于抗结核分枝杆菌时,可协同异烟肼抑制分枝菌酸合成。
三、AI 驱动的虚拟筛选与活性预测
1. 基于机器学习的构效关系(SAR)建模
收集8-羟基喹啉衍生物的抗菌活性数据(如 MIC 值),使用 RDKit 生成分子描述符(如 LogP、氢键供体 / 受体数、拓扑极性表面积),结合随机森林(Random Forest)或梯度提升机(XGBoost)构建预测模型,例如,模型显示 LogP 值在 2.5-3.2 区间时,8-羟基喹啉衍生物对铜绿假单胞菌的膜通透性很好。
2. 深度学习指导的全新分子设计
利用生成式对抗网络(GAN)或 Transformer 模型,以8-羟基喹啉为骨架生成虚拟分子库,通过以下策略筛选高活性候选化合物:
结合能过滤:使用 AlphaFold-Multimer 预测新分子与靶点的结合自由能,优先选择 ΔG<-8 kcal/mol 的结构(较 8-HQ 的 ΔG=-6.5 kcal/mol 提升结合力)。
成药性质评估:通过 PaDEL-Descriptor 计算类药性参数,排除违反 Lipinski 规则(如分子量>500、氢键供体>5)的分子,例如筛选出的 7-氟-5-甲基-8-HQ衍生物,分子量 412,LogP=3.1,兼具高活性与口服生物利用度。
四、动态模拟验证与优化迭代
1. 分子动力学模拟评估结合稳定性
对优化后的8-羟基喹啉衍生物与靶点进行 100 ns MD 模拟,分析:
结合位点构象变化:如在 IMP-1 酶活性中心,优化后的衍生物是否维持与 Zn²⁺的配位键(键长<2.5 Å),以及与关键残基(His234、Cys221)的氢键占有率(>80%)。
溶剂可及表面积(SASA):若衍生物与靶点的 SASA<150 Ų,提示结合口袋埋藏深,不易被溶剂冲刷,稳定性更佳。
2. 基于自由能微扰(FEP)的亲和力精算
对候选化合物进行 FEP 计算,量化取代基对结合能的贡献,例如,在8-羟基喹啉的5位引入溴原子(ΔΔG=-1.2 kcal/mol)较氯原子(ΔΔG=-0.8 kcal/mol)更能提升与 AcrB 的结合力,指导卤代基的良好选择。
五、实验验证与临床转化方向
1. 体外活性验证
通过棋盘法测定优化衍生物与抗生素的联合抑菌浓度(FIC 指数),如7-甲氧基-8-HQ 与美罗培南联用对耐碳青霉烯肠杆菌(CRE)的 FIC 指数<0.5,协同效果优于 8-HQ 原药。
采用蛋白质结晶学解析衍生物与靶点的共晶结构,验证分子对接预测的结合模式,例如,5-氟-8-HQ与 IMP-1 酶的共晶显示,氟原子与 Leu160 形成范德华相互作用,与模拟结果吻合。
2. 临床转化挑战与策略
毒性规避:8-羟基喹啉的金属螯合作用可能引发体内铁、锌离子缺乏,需通过结构修饰降低系统毒性。例如,在喹啉环 3 位引入极性酰胺基团,可减少衍生物与血清转铁蛋白的非特异性结合,肝毒性降低 40%。
耐药性监测:通过全基因组测序分析细菌对优化衍生物的耐药突变位点,设计多靶点协同分子(如同时靶向金属酶和外排泵),延缓耐药性产生。
基于分子对接的 AI 药物设计为8-羟基喹啉的活性优化提供了 “计算模拟 - 实验验证” 的闭环策略,通过精准调控其与靶点的相互作用、金属螯合能力及成药性质,可开发出高效低毒的抗菌增效剂。未来结合冷冻电镜(cryo-EM)解析动态靶点结构,以及利用强化学习(RL)优化设计路径,有望加速8-羟基喹啉类衍生物从虚拟筛选到临床应用的转化进程,为解决细菌耐药问题提供新型分子骨架。
本文来源于黄骅市信诺立兴精细化工股份有限公司官网 http://www.xnlxgroup.com/