
 ronnie@sinocoalchem.com
ronnie@sinocoalchem.com 15733787306
15733787306







8-羟基喹啉的分子轨道计算与电子结构解析
发表时间:2025-11-038-羟基喹啉的分子轨道与电子结构核心特征为“共轭π体系主导前线轨道、羟基与喹啉环的电子耦合影响配位活性”,通过分子轨道计算(如DFT方法)可明确其轨道能量、电子云分布及成键特性,具体解析如下:
一、分子结构基础与计算方法选择
1. 分子结构特征
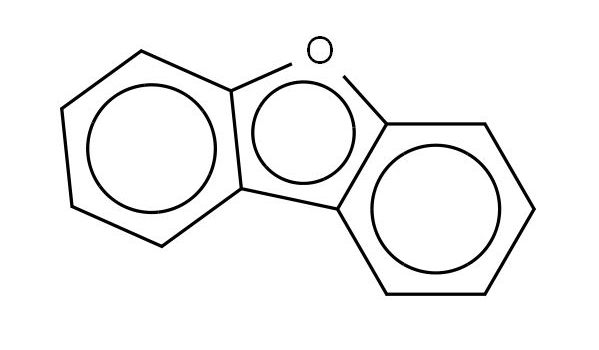
8-羟基喹啉(C₉H₇NO)由喹啉环(含一个吡啶环和一个苯环稠合) 与8位羟基(-OH) 组成,关键结构特点:
喹啉环的6个碳原子(苯环)与4个碳原子+1个氮原子(吡啶环)形成连续共轭π体系,氮原子(N)含一对孤对电子(位于sp²杂化轨道,不参与共轭);
羟基的氧原子(O)含两对孤对电子,其中一对通过p-π共轭与喹啉环的π体系耦合,使O原子的电子向环内转移,增强分子整体共轭性。
2. 常用计算方法
采用密度泛函理论(DFT) 为核心计算方法,结合合适基组可精准描述电子结构,主流参数选择:
泛函:B3LYP(兼顾计算精度与效率,能准确预测共轭体系的轨道能量);
基组:6-311G (d,p)(对N、O等杂原子的价层电子描述更精细,可捕捉孤对电子的轨道特征);
计算内容:优化分子几何构型、计算分子轨道能量(HOMO/LUMO)、电子云密度分布、键级与电荷布局。
二、分子轨道计算结果与电子云分布
1. 前线分子轨道(HOMO/LUMO)特征
前线轨道(至高占据分子轨道HOMO、至低未占据分子轨道LUMO)决定分子的反应活性(如配位、氧化还原),计算结果显示:
HOMO(至高占据轨道):
能量约-5.8~-6.2eV(B3LYP/6-311G (d,p) 水平),电子云主要分布在喹啉环的共轭π体系(苯环与吡啶环均有覆盖)及羟基氧原子上;
成因:羟基O的p轨道与喹啉环π轨道共轭,使O的孤对电子部分融入环内π体系,因此HOMO同时体现“环共轭π电子”与“O孤对电子”的混合特征,这也是8-羟基喹啉能作为配位体(通过O和N原子配位)的关键 ——HOMO的电子云可向金属离子的空轨道转移。
LUMO(至低未占据轨道):
能量约-1.5~-2.0eV,电子云主要集中在喹啉环的吡啶环部分(尤其是与N原子相邻的C原子),苯环部分电子云密度极低;
成因:吡啶环的N原子电负性高于C原子,使LUMO(空轨道)的电子云向N原子附近偏移,导致LUMO具有“缺电子吡啶环”特征,易接受电子(如与富电子基团发生亲电反应)。
2. 其他分子轨道(σ轨道与孤对电子轨道)
σ 轨道:能量较低(<-10eV),主要对应C-C、C-H、C-N、O-H的σ成键轨道,电子云呈“轴对称分布”,键级较高(C-C 键级约1.4~1.6,C-N键级约1.3,O-H键级约0.9),是分子骨架稳定性的核心;
N 原子孤对电子轨道:能量约-7.0~-7.5eV,电子云集中在N原子的sp² 杂化轨道(垂直于环平面),不参与共轭,是分子碱性(接受质子)的来源;
O原子孤对电子轨道:除参与共轭的一对孤对电子(融入HOMO)外,另一对孤对电子位于O原子的sp²杂化轨道(能量约-8.0~-8.5eV),电子云指向环外,可作为氢键供体(与其他分子形成O-H…O/N氢键)。
三、电子结构与分子性质的关联性
8-羟基喹啉的电子结构直接决定其核心性质(如配位能力、溶解性、光谱特征),关键关联如下:
1. 配位活性:O-N双齿配位的电子基础
计算显示,HOMO的电子云在O原子(羟基) 和N原子(吡啶环) 处均有较高密度,且两者在空间上呈“螯合位型”(O与N间距约2.8Å,适合与金属离子形成五元环螯合物);
当与金属离子(如 Al³⁺、Zn²⁺、Cu²⁺)配位时,O和N原子通过HOMO向金属离子的空d轨道转移电子,形成稳定的配位键,这也是 8-羟基喹啉类金属配合物(如Alq₃,有机电致发光材料)的结构核心。
2. 光谱特征:π-π跃迁的轨道根源
紫外-可见(UV-Vis)光谱中,8-羟基喹啉在 240~260nm(强吸收)和310~330nm(弱吸收)的峰,分别对应“π(HOMO)→π(LUMO)”跃迁和“O孤对电子→π(LUMO)”跃迁;
计算得到的HOMO-LUMO能隙(约 3.8~4.7 eV)与实验测得的吸收峰能量(2.8~4.3eV)吻合,证明其光谱特征源于共轭π体系的电子跃迁。
3. 酸性与溶解性:电荷分布的影响
电荷布局计算显示,羟基O原子的净电荷约-0.6~-0.7 e,H原子约+0.4~+0.5 e,使O-H键极性较强,易在极性溶剂(如乙醇、水)中解离出 H⁺,表现出弱酸性(pKa≈9.8);
分子整体呈“极性分子”(偶极矩约3.5~4.0D),且HOMO的电子云分布使分子易与极性溶剂形成氢键,因此在乙醇、DMF等溶剂中溶解度较高,而在非极性溶剂(如正己烷)中溶解度低。
四、计算结果的应用价值
8-羟基喹啉的分子轨道计算与电子结构解析,为其功能化改性与应用提供关键指导:
配位材料设计:通过调控HOMO能量(如在喹啉环引入供电子基团-NH₂,升高HOMO能量),可增强与金属离子的配位能力,优化配合物的发光效率(如用于OLED器件);
药物分子优化:8-羟基喹啉衍生物(如氯碘羟喹)是抗菌药物,电子结构计算可明确其与细菌酶的结合位点(如通过LUMO与酶的富电子基团作用),指导药物分子的结构修饰;
传感器开发:基于LUMO的缺电子特征,可设计8-羟基喹啉基荧光传感器,通过“分析物(如金属离子)与分子结合→改变HOMO-LUMO能隙→荧光强度变化”实现检测。
8-羟基喹啉的电子结构以“喹啉环共轭π体系”为核心,羟基与N原子的电子效应共同调控其分子轨道特征 ——HOMO决定配位与给电子能力,LUMO主导接受电子与亲电反应活性。通过DFT等分子轨道计算,可精准解析其电子云分布、能量水平及与性质的关联,为其在配位材料、药物、传感器等领域的应用提供理论支撑。
本文来源于黄骅市信诺立兴精细化工股份有限公司官网 http://www.xnlxgroup.com/