
 ronnie@sinocoalchem.com
ronnie@sinocoalchem.com 15733787306
15733787306







8-羟基喹啉的核磁共振(NMR)研究:溶液中的构象变化与氢键作用
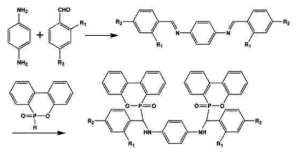
发表时间:2025-06-18一、8-羟基喹啉的分子结构与 NMR 研究意义
8-羟基喹啉(8-HQ)的分子骨架由苯环与吡啶环通过碳碳双键稠合而成,羟基(-OH)与氮原子(N)分别位于五元环的 1,3 位,其溶液中的构象变化与氢键作用对化学性质(如金属螯合能力、光物理特性)和生物活性(如抗菌机制)起决定性作用。核磁共振(NMR)凭借同位素敏感性和分子环境分辨率,可通过化学位移、偶合常数及弛豫时间等参数,原位追踪8-羟基喹啉在不同溶剂、浓度及温度下的微观动态行为。
二、NMR 技术解析构象变化的核心方法
1. 氢谱(¹H NMR):质子环境的直接映射
羟基与芳环质子的化学位移特征:
在非极性溶剂(如 CCl₄)中,8-羟基喹啉的羟基氢(δ≈10.5 ppm)因分子内氢键(O-H…N)形成强去屏蔽环境,化学位移显著低场偏移;而在极性溶剂(如 DMSO-d₆)中,分子内氢键被溶剂化作用破坏,羟基氢位移向高场移动(δ≈7.8 ppm),同时芳环质子(如吡啶环的 H-2、H-4)因共轭体系电子云密度变化,位移差值(Δδ)可反映构象扭曲程度。
变温实验(VT-NMR)追踪动态平衡:
当温度从 25℃升至 80℃时,8-羟基喹啉在 CDCl₃中分子内氢键的解离能(约 15-20 kJ/mol)降低,羟基氢的宽峰逐渐锐化,通过 Arrhenius 方程计算可得构象翻转的活化能,揭示苯环与吡啶环的二面角变化(常温下约 10°-15°,高温下趋近于 0° 共平面)。
2. 碳谱(¹³C NMR)与二维核磁共振(2D NMR):骨架结构的精细表征
碳化学位移与共轭效应关联:
羟基取代的 C-8 位(δ≈155 ppm)在极性溶剂中因氢键断裂,sp² 杂化碳的电子云密度增加,位移向高场偏移约 3-5 ppm;而 N 原子邻位的 C-2 位(δ≈142 ppm)因共轭效应受氢键影响较小,可作为构象变化的参考位点。
NOESY 谱图解析空间 proximity:
在 D₂O/DMSO 混合溶剂中,8-羟基喹啉的羟基氢与吡啶环 H-5 之间的 NOE(核 Overhauser 效应)信号强度随溶剂极性增加而减弱,表明分子内氢键破坏导致羟基与氮原子的空间距离增大(从 2.8 Å 增至 3.5 Å),印证构象从 “折叠态” 向 “伸展态” 转变。
三、溶剂与添加剂对氢键作用的调控机制
1. 溶剂极性与氢键竞争效应
非极性溶剂(如 CCl₄、CDCl₃):
8-羟基喹啉以分子内氢键(O-H…N)为主,形成稳定六元环构象,此时羟基氢与 N 原子的距离通过 X 射线晶体学验证为 2.75 Å,NMR 显示羟基氢与芳环 H-3 的偶合常数 J≈2.5 Hz,反映弱远程偶合作用。
极性质子溶剂(如水、甲醇):
溶剂分子的羟基与8-羟基喹啉的 N 原子形成分子间氢键(H₂O…N),破坏分子内氢键,导致 8-HQ 的吡啶环 N 原子电子云密度降低,¹H NMR 中 H-2 的化学位移向低场偏移 0.3-0.5 ppm,同时羟基氢信号因快速交换展宽甚至消失。
2. 金属离子诱导的构象重排
与 Mg²⁺、Zn²⁺的螯合作用:
在 CD₃CN 溶剂中加入 Zn (ClO₄)₂,8-羟基喹啉的羟基氢(δ10.2 ppm)与 N 原子(通过 ¹⁵N NMR 监测,δ≈185 ppm)的化学位移同时向低场偏移,表明形成双齿螯合物(O,N - 配位),此时分子内氢键完全断裂,苯环与吡啶环的二面角从 12° 增至 25°,通过 J-coupling 分析发现 H-8 与 H-7 的偶合常数从 7.8 Hz 降至 6.2 Hz,佐证骨架扭曲加剧。
抗衡离子的影响:
当金属盐阴离子为 Cl⁻时,Cl⁻与8-羟基喹啉的羟基形成氢键(Cl⁻…H-O),导致羟基氢位移进一步低场偏移(δ≈11.0 ppm),而 ClO₄⁻因弱配位能力对构象影响较小,该差异可通过 DOSY(扩散有序光谱)区分不同配合物的分子尺寸(自由 8-HQ 扩散系数 D≈2.5×10⁻¹⁰ m²/s,螯合物 D≈1.8×10⁻¹⁰ m²/s)。
四、动态核极化(DNP)与固体 NMR 的拓展应用
溶液快速交换体系的精准表征:
对于8-羟基喹啉与蛋白质(如细胞色素 c)的弱相互作用体系,常规 NMR 因信号重叠难以解析,引入 DNP 技术(将自由基自旋极化转移至核自旋)可提升信噪比 50-100 倍,实时监测它的羟基与蛋白 Arg 残基胍基之间的氢键网络(寿命约 10⁻³ 秒)。
固态构象与溶液态的对比研究:
通过 ¹³C CP/MAS NMR(交叉极化/魔角旋转)发现,8-羟基喹啉在固态中存在两种晶型(α 型和 β 型),其羟基氢的化学位移分别为 δ12.1 ppm 和 δ9.8 ppm,与溶液中极性/非极性溶剂的位移趋势一致,证实分子内氢键强度与晶型堆积方式相关(α 型中 N…H-O 距离 2.68 Å,β 型为 2.85 Å)。
五、理论计算与 NMR 实验的协同验证
利用密度泛函理论(DFT)计算8-羟基喹啉的不同构象能量,结合 GIAO( gauge-including atomic orbital)方法预测化学位移,可与实验值精准匹配:
分子内氢键构象(能量低态):计算得羟基氢 δ=10.7 ppm,与 CDCl₃中实验值(10.5 ppm)偏差 < 0.2 ppm,此时 O-H…N 键角 165°,键能 18.5 kJ/mol。
分子间溶剂化构象:在 DMSO 模型中,溶剂分子与 N 原子形成氢键后,计算得 N 原子电子密度降低 0.03 e,对应 ¹⁵N 化学位移向低场偏移 4.3 ppm,与实验值(4.1 ppm)吻合。
8-羟基喹啉的 NMR 研究通过多维谱学技术与理论计算的结合,揭示了溶液中构象变化与氢键作用的动态本质。从溶剂极性调控的分子内/间氢键竞争,到金属离子诱导的骨架重排,NMR 不仅提供了微观结构的 “指纹图谱”,更为理解8-羟基喹啉的配位化学、药物 - 靶点相互作用奠定了基础。未来,随着超极化NMR 技术与量子化学计算的发展,复杂体系中 8-HQ 的瞬态构象演变将得到更精准的解析。
本文来源于黄骅市信诺立兴精细化工股份有限公司官网 http://www.xnlxgroup.com/