
 ronnie@sinocoalchem.com
ronnie@sinocoalchem.com 15733787306
15733787306







公司动态
8-羟基喹啉衍生物的反应活化能分析
发表时间:2026-06-17
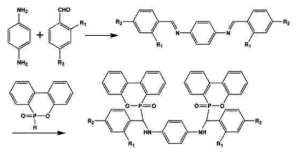
8-羟基喹啉(8-Hydroxyquinoline, 8-HQ)及其衍生物由于具有N,O双配位结构与稳定的芳香共轭体系,在配位化学、有机合成及功能材料领域具有广泛应用。在其衍生化反应与配位反应过程中,反应活化能(Activation Energy, Ea)是决定反应速率、选择性及产物分布的关键动力学参数。
通过对8-羟基喹啉衍生物反应活化能的分析,可以更深入理解其反应机理,并为催化体系设计与工艺优化提供理论依据。
反应活化能的基本意义
反应活化能是指反应物转化为过渡态所需克服的能量屏障,其大小直接影响反应速率常数(k)。
根据Arrhenius方程:
k=Ae−Ea/RTk = A e^{-E_a/RT}k=Ae−Ea/RT
其中:
k:反应速率常数
A:指前因子
Ea:活化能
R:气体常数
T:温度
对于8-羟基喹啉衍生物体系,Ea不仅与键断裂/形成有关,还与分子共轭稳定性及金属配位效应密切相关。
8-羟基喹啉衍生物的结构影响因素
1. 取代基电子效应
不同取代基对活化能影响显著:
给电子基团(–OH, –OCH₃, –NH₂)
o提高电子密度
o稳定过渡态
o降低活化能
吸电子基团(–NO₂, –CF₃, –CN)
o降低电子密度
o过渡态不稳定
o提高活化能
因此,电子效应是调控反应速率的重要手段。
2. 共轭体系扩展
当8-HQ衍生物与稠环结构或芳香基团共轭时:
π电子离域增强
过渡态能量降低
反应路径更加稳定
结果通常表现为活化能降低、反应更容易进行。
3. 分子内氢键作用
8-HQ结构中–OH与N原子之间可能形成分子内氢键:
稳定反应前构象
改变反应路径
影响过渡态构型
可升高或降低Ea(取决于反应类型)
金属配位对活化能的影响
8-羟基喹啉最典型特征是金属螯合能力,在配位反应中活化能变化尤为明显。
1. 金属离子促进反应
与Al³⁺、Zn²⁺、Cu²⁺等金属离子配位后:
提高Lewis酸性中心
增强底物极化
降低键断裂能垒
显著降低Ea
例如Alq₃体系形成过程中,配位反应通常具有较低活化能路径。
2. 配位构象约束效应
金属配位会使分子刚性增强:
限制自由旋转
改变反应路径
可能提高选择性
有时略微提高局部Ea,但降低整体反应能垒
溶剂效应对活化能的影响
溶剂对8-HQ衍生物反应活化能具有显著调控作用:
极性溶剂(DMF、DMSO)
o稳定极性过渡态
o降低Ea
非极性溶剂(甲苯、己烷)
o过渡态稳定性较低
oEa相对升高
质子型溶剂(醇类)
o形成氢键网络
o影响反应路径复杂性
典型反应类型的活化能特征
1. 亲核取代反应
在8-HQ衍生物卤代或酯化反应中:
过渡态涉及芳香体系扰动
Ea中等偏高
受电子效应影响明显
2. 金属螯合反应
典型特点:
配位驱动反应
活化能较低
动力学较快
多为扩散控制或快速平衡过程
3. 偶联反应(Suzuki、Heck等)
在稠环衍生体系中:
需Pd等催化
Ea取决于催化循环步骤
氧化加成步骤通常为控速步骤
计算与表征方法
研究8-HQ衍生物反应活化能通常采用以下方法:
1. 实验方法
Arrhenius图法(lnk vs 1/T)
Eyring方程(过渡态理论)
变温动力学实验
2. 理论计算
DFT密度泛函理论
过渡态搜索(TS optimization)
能量剖面分析
计算方法可揭示反应路径及能垒分布。
应用意义
对8-羟基喹啉衍生物活化能的研究具有重要意义:
优化合成工艺条件
提高目标产物选择性
设计高效催化体系
调控材料性能(光电/发光材料)
指导功能分子结构设计
结论
8-羟基喹啉衍生物的反应活化能受电子效应、共轭结构、金属配位及溶剂环境等多因素共同影响。通过结构调控与反应条件优化,可以有效降低反应能垒,提高反应效率与选择性。未来结合计算化学与实验动力学方法,将进一步深化对其反应机制的理解,并推动其在功能材料与催化体系中的应用发展。
通过对8-羟基喹啉衍生物反应活化能的分析,可以更深入理解其反应机理,并为催化体系设计与工艺优化提供理论依据。
反应活化能的基本意义
反应活化能是指反应物转化为过渡态所需克服的能量屏障,其大小直接影响反应速率常数(k)。
根据Arrhenius方程:
k=Ae−Ea/RTk = A e^{-E_a/RT}k=Ae−Ea/RT
其中:
k:反应速率常数
A:指前因子
Ea:活化能
R:气体常数
T:温度
对于8-羟基喹啉衍生物体系,Ea不仅与键断裂/形成有关,还与分子共轭稳定性及金属配位效应密切相关。
8-羟基喹啉衍生物的结构影响因素
1. 取代基电子效应
不同取代基对活化能影响显著:
给电子基团(–OH, –OCH₃, –NH₂)
o提高电子密度
o稳定过渡态
o降低活化能
吸电子基团(–NO₂, –CF₃, –CN)
o降低电子密度
o过渡态不稳定
o提高活化能
因此,电子效应是调控反应速率的重要手段。
2. 共轭体系扩展
当8-HQ衍生物与稠环结构或芳香基团共轭时:
π电子离域增强
过渡态能量降低
反应路径更加稳定
结果通常表现为活化能降低、反应更容易进行。
3. 分子内氢键作用
8-HQ结构中–OH与N原子之间可能形成分子内氢键:
稳定反应前构象
改变反应路径
影响过渡态构型
可升高或降低Ea(取决于反应类型)
金属配位对活化能的影响
8-羟基喹啉最典型特征是金属螯合能力,在配位反应中活化能变化尤为明显。
1. 金属离子促进反应
与Al³⁺、Zn²⁺、Cu²⁺等金属离子配位后:
提高Lewis酸性中心
增强底物极化
降低键断裂能垒
显著降低Ea
例如Alq₃体系形成过程中,配位反应通常具有较低活化能路径。
2. 配位构象约束效应
金属配位会使分子刚性增强:
限制自由旋转
改变反应路径
可能提高选择性
有时略微提高局部Ea,但降低整体反应能垒
溶剂效应对活化能的影响
溶剂对8-HQ衍生物反应活化能具有显著调控作用:
极性溶剂(DMF、DMSO)
o稳定极性过渡态
o降低Ea
非极性溶剂(甲苯、己烷)
o过渡态稳定性较低
oEa相对升高
质子型溶剂(醇类)
o形成氢键网络
o影响反应路径复杂性
典型反应类型的活化能特征
1. 亲核取代反应
在8-HQ衍生物卤代或酯化反应中:
过渡态涉及芳香体系扰动
Ea中等偏高
受电子效应影响明显
2. 金属螯合反应
典型特点:
配位驱动反应
活化能较低
动力学较快
多为扩散控制或快速平衡过程
3. 偶联反应(Suzuki、Heck等)
在稠环衍生体系中:
需Pd等催化
Ea取决于催化循环步骤
氧化加成步骤通常为控速步骤
计算与表征方法
研究8-HQ衍生物反应活化能通常采用以下方法:
1. 实验方法
Arrhenius图法(lnk vs 1/T)
Eyring方程(过渡态理论)
变温动力学实验
2. 理论计算
DFT密度泛函理论
过渡态搜索(TS optimization)
能量剖面分析
计算方法可揭示反应路径及能垒分布。
应用意义
对8-羟基喹啉衍生物活化能的研究具有重要意义:
优化合成工艺条件
提高目标产物选择性
设计高效催化体系
调控材料性能(光电/发光材料)
指导功能分子结构设计
结论
8-羟基喹啉衍生物的反应活化能受电子效应、共轭结构、金属配位及溶剂环境等多因素共同影响。通过结构调控与反应条件优化,可以有效降低反应能垒,提高反应效率与选择性。未来结合计算化学与实验动力学方法,将进一步深化对其反应机制的理解,并推动其在功能材料与催化体系中的应用发展。